Klinik araştırmalar, üzerinde araştırma yapılacak kimselerin emniyetini sağlamaya ve araştırmanın sağlıklı bir şekilde yürütülebilmesine, takibine ve gereğinde acil müdahale yapılabilmesine elverişli ve araştırmanın vasfına uygun personel, teçhizat ve laboratuvar imkânlarına sahip olan; Gülhane Askeri Tıp Akademisi ve askeri eğitim-araştırma hastaneleri dâhil üniversite sağlık uygulama ve araştırma merkezleri, üniversitelere bağlı onaylanmış araştırma geliştirme merkezleri....
Faz 0 – Preklinik Çalışmalar Geliştirilen ilacın deney hayvanlarında ya da insanlarda mikrodozlar halinde uygulanarak etkene verilen cevap araştırılır.
Faz 1 Çalışmalar; İlacın farmakokinetik özellikleri, toksisitesi, biyoyararlanımı,farmakolojik etkileri az sayıda sağlıklı gönüllüde araştırılır. Bu fazın ana amacı güvenliliktir.
Faz 2 Çalışmalar; İlacın etkili doz sınırları, klinik etkinliği,biyolojik aktivitesi, yarar ve güvenilirliği az sayıdaki hastada araştırılır. Bu aşamada optimum doz ve doz aralıkları hesaplanılır. Bu fazın ana amacı etkinlik ve güvenirliliktir.
Faz 3 Çalışmalar; Birinci ve ikinci aşamayı geçen ilaçlar daha geniş bir populasyonda denenir ve plasebo kontrollü çalışmalarla güvenilirliği, karşılaştırmalı çalışmalarla etkinliği araştırılır. Fazın ana amacı etkinliğin kanıtlanması ve yan etkilerin izlenmesidir.
Faz 4 Çalışmalar; İlk 3 aşamayı geçen ilaçlar ruhsat alır ve pazara verilir. İlaç pazara verildikten sonra yapılan her türlü çalışma 4. faza aittir.
KAVRAM-KANIT ÇALIŞMALARI
Bu klinik çalışmalar hedef nüfustaki ilaç güvenliliğini ölçmek ve doz ile hedeflenen etki arasındaki ilişkiyi, doğrudan ya da dolaylı ölçümlerle araştırmak için tasarlanmıştır.Bu klinik çalışmalar hedef nüfustaki ilaç güvenliliğini ölçmek ve doz ile hedeflenen etki arasındaki ilişkiyi, doğrudan ya da dolaylı ölçümlerle araştırmak için tasarlanmıştır.Bu küçük ölçekli çalışmalarda , ilacın patofizyolojik olarak uygun mekanizmalar ve klinik sonuçlar üzerinde etkili olduğunu göstermek amaçlanır.
KAVRAM-KANIT CALIŞMALARI - II
Kavram-kanıt çalışmaları erken klinik ilaç geliştirme sürecini ifade eder. Faz 1 ve Faz 2a adlı iki döneme ayrılır.Kavram-kanıt çalışmaları erken klinik ilaç geliştirme sürecini ifade eder. Faz 1 ve Faz 2a adlı iki döneme ayrılır. Faz 1; 10-20 kadar sağlıklı gönüllüye tek doz veya kısa süreli tedavi (örnegin 2 hafta) verilmesiyle yapılır. Bu fazdaki calışmaların amacı yeni ilacin istenilen ve fizyolojik olarak tolere edilebilecek bir klinik etkisi olduğunu göstermektir. (Örnegin yeni geliştirilmiş bir hipertansiyon ilacının gerçekten de kan basıncı düşürücü etkisini görmek).Aynı zamanda Faz I, ileriki fazlarda calışılacak olan doz değerleri hakkında rehberlik etmektedir.Faz I calışmalarında bazen de yeni ilacin emilimi, dagilimi, metabolize edilmesi ve atiliminin (ADME) araştırılması hedeflenir.
KAVRAM-KANIT ÇALIŞMALARI - III
Faz IIa; genel olarak ilgili 100 kadar hastadan oluşan grupta yürütülür ve yeni ilacın arzu edilen klinik aktivite icin başarılı olduğu dozu ve uzun vadeli tedavide hastalarin tolere edebileceği uygun dozları belirlemeyi amaçlar (ilacin nihai pazarlama dozu da bu aşamada belirlenir)Faz IIa; genel olarak ilgili 100 kadar hastadan oluşan grupta yürütülür ve yeni ilacın arzu edilen klinik aktivite icin başarılı olduğu dozu ve uzun vadeli tedavide hastalarin tolere edebileceği uygun dozları belirlemeyi amaçlar (ilacin nihai pazarlama dozu da bu aşamada belirlenir) Bu noktada ilacın geliştirilmesi sürecine devam edilip edilmeyeceğine karar verilir. Eğer devam edilirse Faz IIB ve Faz III süreçlerine geçilir.
KAVRAM-KANIT ÇALIŞMALARI - IV
Faz III; daha büyük sayıda hasta gruplarına, pazarlanan kullanım dozlarinda ve sürelerinde randomize olarak placebo ve/veya aktif ilac verilerek karşılaştırılma yapılmasını içerir. Kısa dönemli ve küçük çalışmalara nazaran daha inandırıcıdır ve istatistiksel olarak daha anlamlıdır. İlacın güvenilirliğinin piyasaya çıkmadan önce daha iyi değerlendirilmesini amaçlar.Faz III; daha büyük sayıda hasta gruplarına, pazarlanan kullanım dozlarinda ve sürelerinde randomize olarak placebo ve/veya aktif ilac verilerek karşılaştırılma yapılmasını içerir. Kısa dönemli ve küçük çalışmalara nazaran daha inandırıcıdır ve istatistiksel olarak daha anlamlıdır. İlacın güvenilirliğinin piyasaya çıkmadan önce daha iyi değerlendirilmesini amaçlar.Ilacın etkili ve güvenli olup olmadiğina dair karar bu noktada alınır ve ilacin klinik calışmalar dışında piyasaya sürülebilmesi icin düzenleme yapan otoritelere (FDA ve EMA gibi) başvurusu yapilir.Klinik calışmalar, ilaç markete çıktıktan sonra da; güvenliliğinin daha da belirginleşmesi, ek doz kullanımları ve diğer ilaçlarla birlikte kullanımı gibi konularda devam eder.
Faz 1 (Klinik Farmakolojik Çalışmalar)
Genellikle az sayıda sağlıklı gönüllüde (20-80 kişide) güvenlilik ve toksisite yönünden uygun doz aralığının saptanması amaçlanır. Yaklaşık 3-6 ay süreyle özel birimlerde gerçekleştirilir. Klinik farmakolog tarafından yürütülür.Genellikle az sayıda sağlıklı gönüllüde (20-80 kişide) güvenlilik ve toksisite yönünden uygun doz aralığının saptanması amaçlanır. Yaklaşık 3-6 ay süreyle özel birimlerde gerçekleştirilir. Klinik farmakolog tarafından yürütülür.
Faz 1 çalışmalar bazı durumlarda hasta gönüllüler üzerinde de yapılabilir. Bu duruma örnek olarak toksisitesi yüksek olan kanser ilaçlarıyla yapılan çalışmalar gösterilebilir. İlacın tek dozu ile insanda ortaya çıkan etkiler belirlenirken, kademeli doz artışı ile de farklı dozlardaki toksisite aşamaları izlenmiş olur. Bu fazın ana amacı güvenilirlik araştırmasıdır.
Faz 1’de ki Amaç
- Bu fazın ana amacı “güvenlilik“ tir.
- Tolere edilebilen en yüksek dozun belirlenmesi
- Doz sınırlayıcı istenmeyen etkilerin belirlenmesi,
- Doz sınırlayıcı toksisitenin belirlenmesi (dose limiting toxicity),
- İnsanlarda etki mekanizmasının incelenmesi,
- İlacın insandaki farmakokinetik profilinin gösterilmesi:
- Emilim, dağılım, metabolizma ve atılım düzeyinde inceleme
- Faz II çalışmalar için hastalara verilecek farmasötik şeklin geliştirilmesi
- Faz II çalışmalar için güvenli doz (en iyi doz) aralığının tanımlanmasıdır.
FAZ 1 ARAŞTIRMA TÜRLERİ
Güvenlilik ve tolerabilite çalışmaları
- Tek Doz
- Çoklu Doz
Farmakokinetik Çalışmalar
- Doz proporsiyonalite
- ADME
Özel durumlar
- İlaç etkleşimleri
- Besin etkileşimleri
- Organ yetersizlikleri/Yaşlılar
Gönüllülerin seçilmesi
- Genellikle erkek gönüllüler
- 18-35 yaş arası
- Hepatit & HIV gibi serolojik olayı olmayan
- Alkol bağımlılığı olmayan
- Sigara içmeyen
- Aşırı kafein ve kafeinli içecek tüketimi olmayan
- Enzim polimorfizmleri olmayan
- Stres düzeyi yüksek olmayan
İLAÇ VE BİYOLOJİK ÜRÜNLERİN KLİNİK ARAŞTIRMALARI HAKKINDA YÖNETMELİK
Resmi Gazete Tarihi: 13.04.2013 Resmi Gazete Sayısı: 28617
- Klinik araştırma yapılacak yerler, standartları ve izin başvurusu
- MADDE 11 – (1) Klinik araştırmalar, üzerinde araştırma yapılacak kimselerin emniyetini sağlamaya ve araştırmanın sağlıklı bir şekilde yürütülebilmesine, takibine ve gereğinde acil müdahale yapılabilmesine elverişli ve araştırmanın vasfına uygun personel, teçhizat ve laboratuvar imkânlarına sahip olan; Gülhane Askeri Tıp Akademisi ve askeri eğitim-araştırma hastaneleri dâhil üniversite sağlık uygulama ve araştırma merkezleri, üniversitelere bağlı onaylanmış araştırma geliştirme merkezleri ve Bakanlık eğitim ve araştırma hastanelerinde tercihen klinik araştırma yapmak üzere tasarlanmış yerlerde yapılabilir. (Ek cümle:RG-25/6/2014-29041) Bu merkezler ve hastanelerde yapılan klinik araştırmalara, gereğinde bu merkezlerin ve hastanelerin koordinatörlüğünde veya idarî sorumluluğunda olmak kaydıyla, belirtilen nitelikleri haiz diğer sağlık kurum ve kuruluşları da dâhil edilebilir.
- (2) Faz I klinik araştırmaları ve biyoyararlanım-biyoeşdeğerlik çalışmaları, Kurumun onayladığı, acil müdahale yapılabilmesine elverişli imkânlara ve her biri için ayrı belirlenmiş standartlara sahip, Bakanlık veya üniversitelere bağlı olan sağlık kurum ve kuruluşları ve araştırma-geliştirme merkezlerinde yapılır.
- (3) İyi Klinik Uygulamaları Kılavuzu esas alınarak klinik araştırma yapılacak yerler asgari olarak;
- a) Araştırmanın niteliğine göre gerekli ve yeterli personel ve ekipmana,
- b) Araştırma ürününün niteliğine göre ürünün saklanması ve dağıtılması için gerekli yer ve imkânlara,
- c) Acil müdahale gerekebilecek durumlar da dâhil olmak üzere gönüllü için uygun bakım hizmeti verecek imkân ve donanıma,
- ç) Gönüllünün gerektiğinde daha ileri bir sağlık kurum veya kuruluşuna nakledilebilmesini mümkün kılacak yeterli imkân ve donanıma,
- d) Araştırmanın tamamlanmasından sonra klinik araştırmaya ve gönüllülere ait bilgi ve belgeleri muhafaza edebilecek yeterli imkân ve donanıma,
- (e) (Ek:RG-25/6/2014-29041) Kurum tarafından düzenlenen izin belgesine,sahip olmak zorundadır.
- MADDE 16 – Aynı Yönetmeliğin 26 ncı maddesinin dokuzuncu fıkrası ve onuncu fıkrasının (g) bendi yürürlükten kaldırılmış, birinci fıkrası ile onuncu fıkrasının (b) bendi ve on birinci fıkrasının (b) ve (ç) bentleri aşağıdaki şekilde değiştirilmiştir.
- “(1) Etik kurullar gönüllülerin hakları, güvenliği ve esenliğinin korunması amacıyla araştırma ile ilgili diğer konuların yanı sıra gönüllülerin bilgilendirilmesinde kullanılacak yöntem ve belgeler ile bu kişilerden alınacak olurlar hakkında bilimsel ve etik yönden değerlendirme yapmak amacıyla, üyelerinin çoğunluğu doktora veya tıpta uzmanlık seviyesinde eğitimli sağlık meslek mensubu olan, iyi klinik uygulamaları ve klinik araştırmalar hakkında temel eğitim almış en az yedi ve en çok on beş üyeden oluşturulur.”
- “b) Farmakoloji alanında doktora yapmış veya bu alanda tıpta uzmanlık eğitimi almış kişi,”
- “ç) Biyofarmasötik, farmakokinetik veya farmasötik teknoloji alanında doktora yapmış eczacı,”
Faz 2 Çalışmalar (Faz 1’de sağlıklı gönüllülerde saptanan erken terapötik çalışmalar)
- Faz 2 çalışmalar, 100-200/300 arası hasta gönüllüde,faz 1’de sağlıklı gönüllülerde saptanan tolere edilebilir dozla yapılır.
- Homojen hasta grubu üzerinde ortalama 2 yıl devam eden nispeten kısa süreli çalışmalardır.
- Faz 2 çalışmalarda mutlaka klinik veya tıbbi farmakolog olmalıdır.
Faz 2’de Amaç:
- Bu fazın ana amacı ‘etkinlik ve güvenlilik’ belirlenmesidir
- Hedef hasta grubunda ilacın terapötik endikasyondaki etkinliğini
- Etkili optimal dozu (terapötik doz aralığı),
- Yan etki profilini,
- Güvenliliğini anlamaktır
Bu çalışmalar Faz 3 çalışmalarının fizibilitesini ortaya koymak amacıyla kontrollü, randomize veya çift-kör çalışmaya dönüşürse Faz 2b (geç Faz 2) olarak adlandırılır.
Faz 3 (Yaygın Terapötik Çalışmalar)
- Faz 3 çalışmalar, 1000-3000 hasta gönüllüde üç dört yıl süren, genellikle çok merkezli, çok uluslu, randomize, çift-kör nitelikteki çalışmalardır.
- Resmi kuruluşa yeni ilaç başvurusu yapılana kadar ürünün klinik etkinliğinin ve yan etkilerinin daha geniş bir hasta popülasyonunda değerlendirilmesi söz konusudur.
Faz 3’te Amaç:Faz 3’te Amaç
- Ana amaç: “etkinliğin kanıtlanması ve yan etkilerin monitörizasyonu”dur.
- Bu fazda etkililik ve güvenlilik bakımından tedavi amacıyla kullanılan diğer ilaçlarla kıyaslama yapılabilir.
Ürün Onayı:
Faz 3 çalışmalarda yeterli veriler elde edildikten sonra ürünün ilaç olarak kullanılabilmesi için onay alınması gereklidir.Faz 3 çalışmalarda yeterli veriler elde edildikten sonra ürünün ilaç olarak kullanılabilmesi için onay alınması gereklidir.
Türkiye’de yeni araştırma ilacı ile ilgili klinik çalışma ve ruhsat onayı Sağlık Bakanlığı İlaç Eczacılık Genel Müdürlüğü tarafından verilmektedir.Avrupa Birliği’nde “European Medicines Evaluation Agency (EMEA)yeAmerika Birleşik Devletleri (ABD)’nde “Food and Drug Adminstration (FDA)”a, “New Drug Application (NDA)”a başvurulması gerekmektedir.
Ürün Onayı II
- Onay süresi yaklaşık 1-1.5 yıldır.
- ABD’de FDA tarafından özellikle yeni onkoloji ilaçları ve mevsimsel çalışmalar için ‘Fast Track (hızlı geçiş)’- hızlı onay işlemi uygulanabilmektedir.
Faz 4 (Pazarlama Sonrası Çalışmalar)
- Ruhsat almış bir ilaç ile yapılan geniş amaçlı pazarlama sonrası izlem çalışmalarıdır. İlacı kullanmakta olan geniş hasta toplulukları söz konusudur.
Faz 4’te Amaç
- Ana amacı “uzun süreli güvenilirlik” verilerinin toplanmasıdır.
- İlacın kullanımının ekonomik boyutlarının incelenmesi,
- İlacın maliyet-yarar-risk oranlarının analiz edilmesi,
- İlaçla ilgili cevaplanmamış soruların cevaplanması,
- Faz 3’te başlamış olup tamamlanmamış olan çalışmaların tamamlanması,
- Yeni ilacın etkinliğinin daha geniş hasta gruplarında gösterilmesi,
- Ender rastlanan ciddi advers olayların ve ilaç etkileşimlerinin belirlenmesi,
- Yeni tedavi endikasyonları için ipuçları sağlanmasıdır.
Etik Kurul Onayı
- Faz 1, 2 ve 3 çalışmaları için Yerel Etik Kurul onayı ve bakanlık izni alma zorunluluğu vardır.
- “Ruhsatlı ilaçların kabul edilen endikasyonları üzerinde emniyeti, doz farklılıkları ve yan etkileri gibi konuları araştıran çalışmalar’ olarak tanımlanan Faz 4 çalışmalarının yapılabilmesi içinse sadece Yerel Etik Kurul onayı yeterlidir.
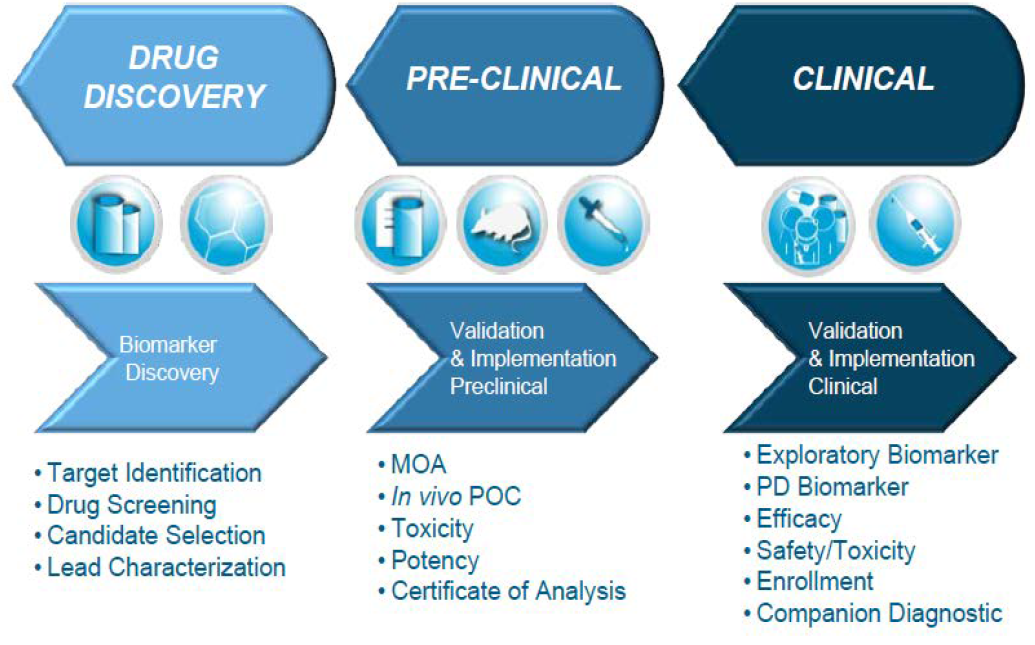
Biological Marker (Biyobelirteç)
- Normal biyolojik süreçlerin
- Patolojik süreçlerin
- Terapötik müdahaleye farmakolojik cevapların değerlendirilmesinde objektif olarak ölçülen, karakteristik göstergelerdir.
Amaç nedir?
- Klinikte hastalık için risk belirlenmesi
- Hastalığın teşhisi
- Hastalığın şiddeti
- Tedavi stratejilerinin belirlenmesi
- Hastanın tedaviye cevabının izlenmesi
AVANTAJLARI NELER?
- Klinik araştırma ortamında kullanıldığında, biyobelirteçlerle yapılan çalışmalara göre ilacın güvenli ve etkin olup olmadığı gözlemlenip, klinik sonuçlar daha kısa zamanda ve daha düşük maliyetle tahmin edilebilir.
- Erken teşhis olanağı sağlar
- Çalışmaların efektif ilerleyip doğru tedaviye yönlenmesini sağlar
- Yeni ilaç araştırılmasında faydalanılır
- Kişiselleştirilmiş tedavide gelecek vaadetmektedir
- Prediktif biyobelirteçlerin kullanılabilirliği ile hasta popülasyonunda yapılan sınıflandırmalar klinik araştırmaların daha küçük popülasyonlarla yapılabilme kolaylığını sunmaktadır.
- Bütçelerin daha verimli kullanılmasını sağlamaktadır.
- Toksisite biyobelirteçleriyle olası yan etkilerin belirlenmesi sonucunda, klinik araştırmaların sayısı ve süresi azalmakta ve bu azalış ile araştırmalar için harcanan maliyet düşmektedir.
Uygunluk Araştırması
Bir biyobelirteçin uygunluk araştırması; biyobelirteçin hastalıkla biyolojik bağlantısı veya hastalığın klinik sonuçlarıyla olan bağlantısının ispatlandığı süreçtir. Bir biyobelirteçin geçerliliğinin onaylanması; biyobelirteçin karakteristik performans kalitesinin ve özgüllüğünün ölçülmesi için yapılan tahlil ve ölçüm teknikleri sonucu olur.
Klinik Biyobelirteç Örnekleri
- Proteinler ve peptidler (örn; PSA )
- Antikorlar (örn; RA)
- Hücreler (örn; bazı kanserlerde lökosit),
- Metabolitler (örn; FKU için idrarda fenilalanin),
- Lipitler (kardiyovaskuler hastalık),
- Hormonlar
- Enzim düzeyleri (HCC’de KCFT),
- RAI ( Tiroid fonk.)
SAFE-T (Safer and Faster Evidence-based Translation) -Daha Güvenli ve Daha Hızlı Kanıta Dayalı Çeviri
- SAFE-T çalışma, kamu sağlığını iyileştirmek ve kişiselleştirilmiş ilaç tedavisinin gelişimini sağlamak için yaratıcı çalışma yöntemleri oluşturmayı hedeflemektedir.
- İlaç geliştirmede önemli zorluklardan biri hasta güvenliğidir. Sıklıkla birçok ilacın yan etkileri öngörülebilir değildir. Yüksek riskli durumlarda ciddi yan etkilerin geç tespiti sorun yaratmaktadır. Yan etkiler teşhiste ve ilaç geliştirmede önemli bir engeldir.
- SAFE-T projesi dahilinde hastaların kan ve idrarlarındaki belirteçler kullanılarak; ilaçlardan kaynaklanacak üriner, hepatobiliyer ve hematolojik yan etkilerin öngörülebilirliğinin artırılması amaçlanmaktadır. İlaç kaynaklı hasarlanmaları izlemek için duyarlı ve spesifik klinik testlerin mevcut eksikliği giderilmeye çalışılıyor.
- SAFE-T konsorsiyumu bu üç sistemin her biri için, daha spesifik, daha duyarlı ve şu anda mevcut olanlardan daha akıllı biyobelirteçleri tanımlamak, ve bu biyobelirteçlerin rutin kullanımını sağlayacak düzenlemelerin kabul edilmesini sağlamak için çalışmaktadır.
- Bu amaca ulaşmak için, konsorsiyum Avrupa ve ABD'de düzenleyici kurumlar ile yakın işbirliği içinde çalışarak ve başka konsorsiyumlar ve araştırma grupları ile bağlantılar kurmaktadır. Konsorsiyum tarafından toplanan numuneler merkezi bir veri bankasında 5 yıl süreyle saklanır.
BİYOBENZERLER
- Biyobenzerler, aktif maddesi rekombinant DNA ya da kontrollü gen ekspresyon yöntemleri vasıtasıyla canlı bir organizma tarafından yapılan veya türetilmiş biyolojik tıbbi ürünlerdir.
- Biyobenzer ilaçlar halihazırda kullanım yetkisi alınmış olan başka biyolojik ilaçlara benzediği için bu isimle anılmaktadır.
- Referans ilaçla kalite, güvenlik veya etkinlik açısından anlamlı bir farklılıkları yoktur.
- Referans ilaç hakkında güvenlik ve yetkilendirme bilgileri zaten mevcut olduğundan, biyobenzerin etkinliği hakkında gereken bilgi miktarı orijinal bir biyolojik ilaç için gereken miktardan daha azdır.
- Biyolojiklerin yüksek maliyetli üretim süreci nedeniyle, hızlandırılmış onay süreci avantajı biyobenzerlerin Avrupa’da önemli bir pazar payı kazanacağını düşündürmektedir.
- FDA 23 Mart 2010 tarihinde Başkan Obama tarafından imzalanan Hasta Koruma ve Ekonomik Bakım Yasası kapsamında biyobenzerlerin onaylama yetkisini kazanmıştır.
RUHSATLANDIRMA ve ÜRETİM
- Biyobenzerler ancak orijinal referans biyolojik ilaç verilerinin gizlilik süresi dolduktan sonra ruhsatlanabilir. Genel olarak, bu demektir ki, biyolojik referans ilacın en az 10 yıldır ruhsatlı olması gerekir.
- Avrupa Birliği 2009-10 döneminde toplam biyobenzer satışını 200 milyon dolar olarak belirlemiştir.
- Ancak onay sürecinin yasal zorlukları ve üretim sürecinin maliyeti biyobenzerlerin geliştirilme masrafını da arttırmaktadır; maliyet molekül başına 75-250 milyon dolara kadar çıkabilmektedir.

